1、申报资料
Q:(山东RAX):请问药品质量标准的书写有更新的明确要求吗?CDE发的模板中没有详细的要求。
A:(RA-郭星星):药典委的《关于规范提交化药新药试行标准格式的说明》说明了格式体例要求,以及《国家药品标准(化学药品)正文各论编写细则》讲清楚了内容怎么写。当然内容一般情况下就抄药典。至于一些合理的放宽,或者描述的修饰,就看你注册检验项目经验了。
2、参比制剂
Q:(Mr.Z):请教各位老师,5.2类注册,关于参比制剂的选择,有要求一定选择公布的国内上市的进行质量对比吗?是否有要求选择参比制剂进行稳定性放样研究呢?
A:():参比制剂稳定性见CDE一般性技术问题解答第4条:未强制要求质量对比:要么选择国内参比制剂,要么备案你们选择的国外上市。
3、注册检验
Q:(南京-注册-小e):想咨询下 关于批准后的药品补充申请时发补的注册检验,去省所注册检验的时候,那个自检报告,还需要按照修订后的质量标准再检验一次吗?
A:(江西-注册-Tim):需要。发补时的注册检验,必须按修订的质量标准检验一次。发补通知中,让你们是全检,还是部分项目检验。如果是全检,就必须按修订后的质量标准全检并提供报告单,不需要再提供按修订前的报告单。
4、光盘申报
Q:(中美双报~人药~heheng):请问一下最近递交光盘申报的老师,你们光盘里是按照最后分册装订的形式去交的吗?还是就按照ICH M4形式刻录进去的?譬如这种
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(沪-打杂-���):我们都是按照M4里面的粒度文件 比如P1 P2 这种。刻盘。
A:(注册er):2020-07-09 药品注册申报资料格式体例与整理规范 (2020年第12号)
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
5、法规政策
Q:(紫韵心情-注册-河北石家庄):想请教大家一下,对于原来中药品种化药文号的品种,增加规格或改剂型,现在新的政策是怎样的?
A:(四川-研发-笨笨熊):按化药管理,今年目前没有新政策
6、资料申报
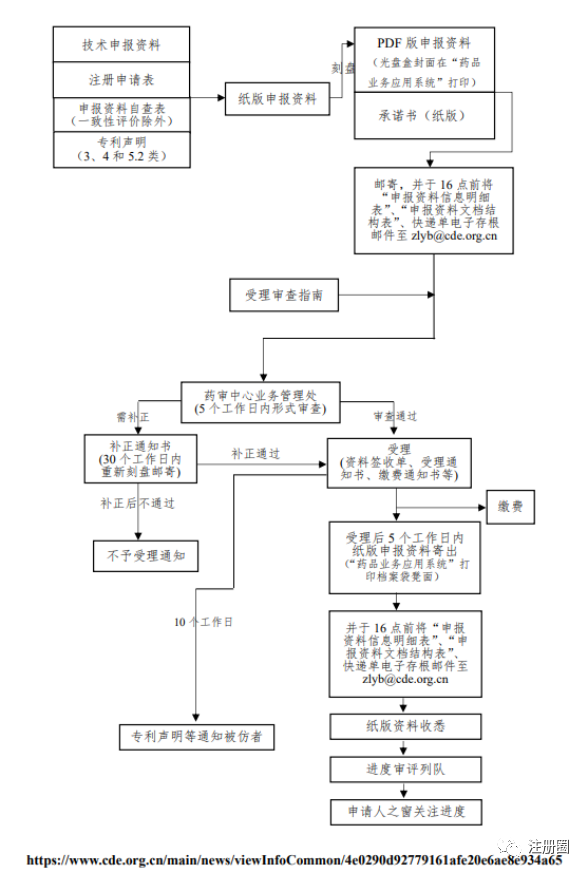
Q:(广东注册Chloe):我想知道新的受理流程小白老师,能不能也给我们分享一哈?
A:(沪-打杂-���):不知道对不对
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
总结下来就是:1.只要寄东西给cde,即使只有一张纸,也要发邮件2.新报要先光盘后纸版3.补充申请等按现在的,直接纸版
7、专利声明
Q:(豫小南):原料药模块一“1.3.8.2 专利信息及证明文件”,该怎么提供,我们之前都是提供一个声明,现在不知道还可行不?
A:(四川-研发-笨笨熊):你在那个网页查下别人家已经公示的专利声明,按那个格式
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
8、资料递交
Q:(沪-打杂-���):境内低风险地区寄给cde的资料要核酸报告么?附件二里面那里怎么填,未进行检测?A1:(南京-注册-张nn):第一个问题,不需要。
A2:(北京-RA-Lynn):第二个问题,不适用。
9、申报资料
Q:(北京-刘蔚佟(Chris)- eCTD):各位老师,临床试验数据是否也需要跟着光盘交,还是只跟着纸质资料一起交?
A:(沪-打杂-���):放在纸版资料第一袋,第一套第一袋。
10、申报资料
Q:(山东-注册-Jessie):有没有老师知道,这个是填什么编号
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(山东-注册-Jessie):流水号。申请表右上有两个编号,一个是流水号一个是核对码。
A2:(中美双报~人药~heheng):这个页面受理老师好像看不到,是企业看到的。我们打电话问了受理,确实填流水号。这样不同规格的核对码不同,但流水号一样的了。第一个选择否,第二个规格填的时候选择是。系统上显示两个不同规格的流水号一致,意思就是这两个不同规格关联的一套资料,年后才有这个选项的。
11、申报策略
Q:(沪-打杂-���):各位大神:有个国内特有规格A,参比制剂B规格,想报个B,怎么操作比较好?一致性评价?还是新4?我觉得按一致性评价报B比较好
A1:(深圳-注册-tjw):个人意见:先做BE增加规格补充申请,用一致性评价的申请表,注明走一致性评价序列,可能获批后,该规格视同通过一致性评价。依据: 257号文件:(一)属于下列情形的化学药,应当进行BE试验备案:1.仿制已上市的参比制剂,其活性成分、给药途径、剂型、规格应与参比制剂相一致。参比制剂应为原研药品。2.已批准在境内上市,需通过BE试验开展相应变更研究的药品。
CDE一般性技术问题:20210608:增加药品规格的补充申请,能否合并一致性评价申请一同申报?答复:一致性评价申请按品种管理,开通新增药品规格的补充申请直接申报一致性评价途径。为了防止规格滥用,新增加的药品规格需符合临床合理用法用量范围,且与原研(参比制剂)一致。
A2 注册圈):如果原规格A 与拟增加规格B 为同一持有人,按A1回答操作没问题;如果规格A 是其他家持有,现在自己想做规格B,按新4比较合适。 注册圈):如果原规格A 与拟增加规格B 为同一持有人,按A1回答操作没问题;如果规格A 是其他家持有,现在自己想做规格B,按新4比较合适。
12、注册批
Q:(nothingོ):请问注册批可以是中试规模生产的嘛?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(沪-打杂-���):那你商业化规模就得是中试规模
A2:(四川-研发-菜鸟):必须要商业化规模批次
A3:(nothingོ):要在注册批生产规模符合要求的前提下
13、杂质控制
Q:(注册):请教大家:标准中已知杂质限度是0.2%,参比制剂裸样放光照没有,但是我们自制制剂裸样放光照5天没有(报告限以下)、光照10天产生了(含量0.1%),就是说裸样时自制制剂(小试产品没有、两批中试产品都有)产生了一个杂质,参比制剂无。包装后的自制制剂没产生是否可行呢?这个是光降解杂质,带包装考察的自制制剂没有这个杂质。小试样品(烧杯配制)都没有,中试(不锈钢配液罐)放大2批都有这个杂质。参比制剂裸样光照没有产生这个杂质,我们本来下一步要进行工艺验证了,现在不知道怎么办。我们这液体制剂一上中试就出问题,厂家设备都是不锈钢的,我们还都是pH3-4左右的品种。
A1:(长沙-注册-蛋蛋):我们有个品种,差不多类似的情况,影响因素光照下辅料含量降低,但是参比光照未降低,避光条件下自研和参比辅料都不降低,最后分析出来是原料药的金属离子导致的,CDE要求把原料药的金属离子做了严格控制。你这个是杂质变化。
A2:(上海-注册-贰雯):你们还是先调查清楚杂质来源,再说解决办法。如果解决办法可靠,这个数据上审评是没有关系的。
A3 :(注册圈):建议排查配液罐搅拌桨是否有金属离子引入,有些品种在一定pH值条件下,对金属离子比较敏感,会催化杂质产生。我们有个品种在做液相检测杂质时,杂质时有时无,排查找到原因是色谱柱的保护柱管套有金属离子引入液相
14、IND申报
Q:(云):圈友们,IND申报到底要不要提交模块五呐?记得之前群里有老师说过要交,今天有个老师告诉说IND不需要模块五,NDA才需要。有点蒙圈呐
A1:(北京-药品注册-G):要的啊,只是IND阶段没有临床试验结果的,相关章节下写不适用或未开展相关研究,NDA的时候需要提交详实的试验数据;如果不提交,形式审查的时候就会卡住吧?另外,撰写临床方案和IB涉及的参考文献,我们也是放在模块5的
A2:(上海-RA-冯新宇):IND不用交啊 IB 放在1.3.4就好了,IND 我们模块五写的不适用
15、工艺验证
Q:(deardeerreed-注册-上海):大家好,请教一个问题。对于不同规格的制剂产品(处方完全等比,前面制粒总混完全一样,只是最后灌装量不一样),在各做3批工艺验证的时候,可以只做一个规格的全验证吗?另一个规格只从灌装开始做验证?
A:(上海-注册-贰雯):问了几个质量体系下的大佬,反馈:新产品首次验证最好都要全过程验证,之后再验证可以考虑用矩阵法或者评估减少验证批次数。——仅供参考
16、工艺验证
Q:(注册):请教下大家:安瓿瓶装的滴眼液或者雾化吸入溶液(无菌工艺)的检漏设备在工艺验证时没有到位,先生产样品后面等检漏设备到位后再检漏可行呢?
A:(长沙-注册-蛋蛋):我们注射液是这么做的,没有问题。申报时可以用传统的色水法检漏,最晚在发补提交时完成确定性的密封性检漏方法研究和确定。
17、光盘申报资料
Q:(江苏 注册 点滴的阳光):请问按照现在电子光盘提交资料要求,申报资料中3.2.P.1这种文件命名中的“.”不允许使用吗?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(北京-药品注册-Rae):我们都去掉了
A2:(RA-上海):不可以的,可以参考ICH的法规里的文件和文件夹名,中文有翻译件。M8里eCTD specification 找找。
18、参比制剂
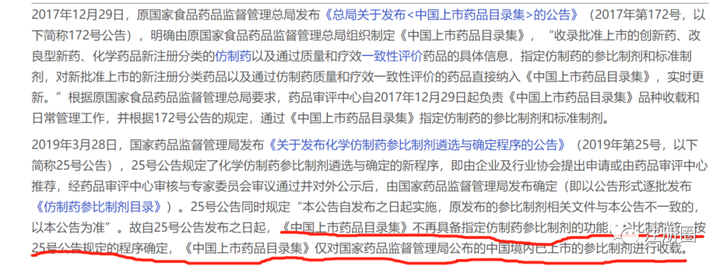
Q:(广东-化药-小榭):请教一下:上市药品目录集收载的标明为RLD的制剂是不是直接就可以作为参比制剂使用,而无需再次对该本品提出参比制剂备案?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
上市药品目录集中已被收载的境内已上市的参比制剂如果参比制剂目录中没有公示,还需要再走一次备案?A:(上海-注册-贰雯):是的,受理那边只认参比目录那些公告。申请表、自查表那些文件,也都需要填写参比在参比目录上对应的序号。
19、工艺验证资料
Q:(北京-注册-项管小白):请教一下各位老师,申报资料中的工艺验证资料可以是研发部门提供的资料吗?产品开发各个阶段由哪个部门负责有相关法规要求吗?
A1:(长沙-注册-蛋蛋):工艺验证应该在GMP体系下进行,一般由工厂提供。
A2:(上海-注册-贰雯):工艺验证要在GMP体系下进行,具体可以看下2010版GMP对工艺验证的各种要求。申报资料的工艺验证都得是转移到生产部门在GMP体系下完成。相当于走三遍商业化生产,证明工艺稳定可行可重现。
20、临床试验
Q:(云):请教圈内的老师们,请问何为:探索性临床试验?何为:关键性临床试验呢?谢谢
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(南京注册丽敏):探索性临床试验一般指2期,关键性临床试验是获批相关的试验,对新药来说,一般指代3期。
21、技术转移
Q:(注册-北京):请教下大家,产品技术转移的具体要求有相关文件依据吗?
A:(上海-注册-贰雯):可参考,2010版GMP-质量管理体系,3.5产品工艺管理,这个章节。2010版GMP指南 质量管理体系。还有 2010版GMP指南-口服固体制剂 5.1.3技术转移 。分析方法,参考药典9100通则,和 2010GMP指南-质量控制实验室 10.3节。再还有个 ICH Q10。
22、除热原验证Q:(黄婷):请教大家一个问题,大输液100ml、250ml输液瓶除热原验证(没有适合的隧道烘箱)该怎么做?是将内毒素标准液涂抹在瓶中内壁上,风干后按照正常清洗程序洗涤吹干,然后检测,看内毒素是否下降3个LOG值吗?谢谢大家!
A1:(长沙-注册-蛋蛋):这样研究的话,还要补一个提取率。
A2:(上海-注册-贰雯):还要确认洗之前的内毒素含量,和理论值差异多大。你这种清洗方式,降低3个对数值,挺难的。为什么不直接买免洗的。我去问了下质量的大佬,他们说这种清洗程序可以降低3个对数值,验证方法也是你说的那样。然后“另外,还要增加最后一次的清洗水的内毒素检测。”回复供参考。
23、 一致性评价
Q:(山西-注册-文璐):各位前辈,一致性评价目前还是按照这个受理吧?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(注册圈 空空):是的。
24、申报策略
Q:(天津-注册mm):请教个问题,属于临床价值明确,无法推荐参比制剂的化学药品,这个里面的产品,大家有没有申报的啊?我看现在还是征求意见稿,是还需要等等吗?
A1:(浙江-注册-小丽):去年有申报过的,目前没有受理。参比备案上去了,也不会同意的。得到的回复是,等待后续相关文件的出台.
A2:(广州-仿制药注册-Jay):前两天出的受理指南征求意见稿把无法推荐参比制剂的申报路径都删掉了,我个人猜3月份会出无法推荐参比制剂的正式稿
.25、场地变更
Q:(深圳+注册+石头):请教各位老师,变更生产场地(持有人自行生产变更为委托生产),主文件清单需要分别提供委托方和受托方所有信息吗?A:(flora):场地主文件是提供生产场地的,委托和受托方要提供变更后的生产许可证.
26、变更类型
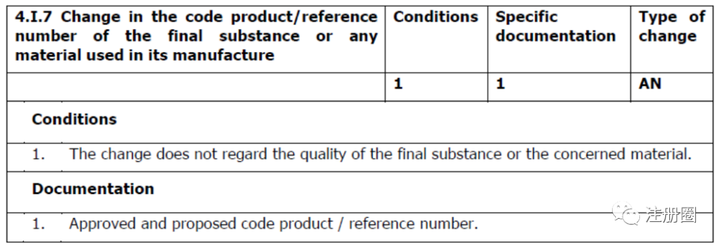
Q:(yydy 11得1):各位老师,有个API老产品,之前申报CEP时定义的1个起始物料,现在发现它不适合当起始物料,想将它降为普通物料,请问是哪种变更?以下变更等级合适吗?谢谢。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(Cynthia Lynn):重新定义的话需要说明原来的降为普通物料,还要重新定义起始物料,对其进行控制和杂质分析,您可以看看变更指导原则质量/物料部分
27、参比制剂申报
Q:(安徽-注册-微子頲):各位老师好,药审中心这句话删除什么意思啊 那意思以后这种类型的没有渠道申报了?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(mshy):也不是没有途径了,有余地,看3.1.2。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
28、 参比制剂
Q:(北京—RA—大卫):请教下各位老师,有没有哪个法规对参比制剂的购买来源有相关的规定的呀?除了这个2018年国家局发布的问答外,不知道还有没有更新的.
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(北京-PM/RA-Frank):就这,没别的,申报资料要求(80号文)里也就这些,不用纠结。目前一个普遍做法就是,上人的(做BE做临床)要拿到一次性批件,不上人的,有说明性文件即可。但一次性批件在省局办也越来越方便,现在倾向于全部用一次性批件进口。
A2:(杭州-红豆):新出的化药受理指南(征求意见稿)中明确写出来了,“2、参比制剂来源证明文件一般包括:官方网站的批准信息,所购参 比制剂的实物和包装标签(包括内包装和外包装)照片,购买发票、 说明书、进口药品批件和进口药品通关单(采用国外上市的参比制剂 时需提供)复印件等相关文件。并同时说明参比制剂的来源(上市国 家或地区)、药品通用名称、英文名、商品名、规格、剂型、持证商 等信息。”
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
29 、年报
Q:(Hailey海燕)各位老师,请问知道药品管理法中的年度报告是怎么提交的吗,是提交到哪个系统啊?
A:(北京-药品注册-Mr.好先生):目前了解到的,国产制剂没有提交通道,省局让先写着,有通道了,再提。有的省局 让企业把 纸件提交给分局备案,有的省局让自己存放。
30、持有人变更
Q:(山西-质量-clf):请教各位老师,我们想买一个产品做持有人,目前这个产品的工艺验证是完成3个月稳定性,还在继续考察,我想问是等这个产品申报之后买呢?还是现在就买过来委托原单位继续考察比较合适?这个有时间上的要求吗?我们想现在买过来,免得以后要单独去做持有人变更,我想咨询一下这个时间上应该啥时候签转让协议比较合适?
A1:(石家庄-药品注册-千千阙歌):单纯的持有人转让不需要稳定性资料,没有关系,先后都没有问题。持有人变更,最好作为一个单独的注册程序去办理,因为是必须要报国家CDE的,是一个非技术审评程序,很简单;不建议和其他变更关联申报.持有人变更注册时一般不会变更生产场地,所以只是单纯的持有人变更,时间无所谓,都不影响。
A2:(注册圈):如果生产企业没有改变,只变更持有人,“等这个产品申报之后买”和 “现在就买过来委托原单位继续考察”都可以,时间上没有要求,建议采用后者。如果持有人和生产企业都改变,建议前者,获批后再走持有人变更和生产场地变更。
31、药典标准
Q:(天天):想请教一个问题,中国药典对“在干燥处保存”的具体要求是什么啊?比如相对湿度的要求?
A:(ZL-制剂-长春)USP里有要求,我搬运过来的,仅供参考。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
32、工艺验证
Q:(小赵-天津注册):请问,工艺验证批生产,需要三批制剂对三批API吗?报国内
A:(成都-注册-蔓蔓妈):可以2对3,最好是1对1。
33、申报资料
Q:(���):各位老师,请问多个包装规格是提交一套申报资料么?如果可以,那是提交一套申报资料,给多个批准文号么?我看已获批的产品,不同包装规格是对应不同批准文号的~
A:(石家庄-注册-朵拉):一套资料一个文号,多个规格。
34、检验报告
Q:(简):请问一下 模块一 1.3.6 检查相关信息需要附检验报告,是什么批次的检验报告呢?
A:(丽珍):注册批和be批。
35、关联申报
Q:(夏天):请问,如果原料药和制剂关联申报,原料药被发补往前延伸一步,重新提交工艺验证资料,那制剂会受啥影响?我们自己的原料药+制剂。
A1:(北京-注册-刘成林):按道理来说,制剂审评应该是等着原料药的补充资料交上来之后再审评。
A2:(北京-药品注册-Mr.好先生):制剂一般会在发补通知中要求,制剂企业关注原料药的工艺变化。你们先评估一下变更前后API的质量有没有变化,然后自然就得出对制剂的影响了呀.如果API 没啥影响,那对制剂的影响是微小的。A3:(注册圈):制剂至少需要提交原料变化前后对制剂影响的风险评估报告及针对不同等级风险采取的控制措施。
36、工艺验证
Q:(浙江-RA-刘玲):各位老师,请教下新药的工艺验证批次能用于III期临床研究吗?
A:(左左右右):GMP条件下生产的就可以。
37、说明书更新
Q:(北京-注册-wolflee):请教一个进口产品说明书安全性问题的更新,如果与该安全性问题相关的适应症在国内并没有批准,那这个安全性信息是否还需要在国内的说明书中更新?比如是美国,那FDA批准那药品的适应症更新了国外说明书,然后按照FDA批准的适应症再更新国内说明书,但那个适应症在国内并没有批准.实际情况是我不打算在国内增加该适应症,但国外出现与该适应症相关的安全性信息(PV评价的结果),国外已更新说明书,现在的问题是:国内的信息要不要同步?
A:(一颗迟梓新):这种增加适应症要申请验证性临床的吧,毕竟有种族人群差异,如果只是药理毒理、禁忌、不良反应等并非与适应症直接关联安全性内容的内容应该可以申请根据原研说明书进行修订。
38、补充申请
Q:(王萌声^0^):请问各位老师一个问题,药品上市前做补充申请吗?为什么有的药品没有上市,就已经有补充申请的受理号了第四位(B)。
A:(长春-研发-melon):上市前可以报补充申请.一般官方不会要求,自己报.没有法规规定只能上市后报,法不禁止即可为,比如临床试验期间的补充申请,可以百度一下。
39、Pre-IND申请
Q:(杭州_注册_sunny):小白请教一下各位大佬,申请创新药的FDA pre-IND,需要像国内这样先注册账号吗?还是直接发送邮件就可以呢?相关邮箱号怎么找呢?
A:(上海- RA-艾兰兰):参考FDA网站的这个文件,可以paper submission或者Gateway,邮件不可以.
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
40、生产许可证办理
Q:(wsf):请问各位老师,如果委托生产B证和C证是同时办理,还是有先后顺序?我看到江苏b证和c证的申请是同步的。
A1:(王冬生):北京,先办C证,否则B证缺材料.场地是受托方的,必须要受托方现场核查确认具备条件省局同意才可以办B证,我们前段时间咨询过,正在办,受托那边已经办完同意受托了。B证企业我还不清楚要不要来现场,C证那边是省局去人查了生产条件,结论就是具备受托生产条件,同意企业受托生产.北京局的意见是没有受托企业那边监管部门的意见,他们没法把握委托生产是否可行。
A2:(注册圈):不同省局处理这类问题的方式不同,安徽和江苏一样,这类地方局办理件最好咨询省局。
41、注册检验
Q:(广东-注册-曾):请问下各位,上市后变更的补充申请受理时,会同时开具检验通知单么?
A1:(浙江-注册-关耳):我们没有开,是发补的时候要求我们检验的。
A2:(山东-注册-三碗不过冈):受理时没有,我们是发补时让检验的。
42、沟通交流会
Q:(在海一边-注册-连云港):想请教有提交过II/III沟通交流会议的吗?该阶段递交研究材料对应的技术要求是按照《创新药(化学药)III期临床试验药学研究信息指南》吗?沟通交流药学相关的关键问题,如工艺路线是否合理等。
A:(董):直接写明你的问题,想这么做的相关依据就可以了。仅提供关键问题及相关支持性资料,其他模块的资料不需提供,如果你的问题不是很多,可以不通过很多资料来描述证明的话,就先写个一般性问题咨询呗.你简单问题,他们不一定会跟你开会的.对,多种方式可选,没必要一定开会,说不定是书面回复。
43、动态核查
Q:请问近期有接受过生物药动态现场核查的吗?请问现在生物药动态核查,一般要求几个批次?
A:(雄_出没):这个可以沟通的,但需在核查阶段能够看到完整的工艺流程,特别是关键的工艺步骤.
44、供应商审计
Q:(璐璐常):请教老师们个小问题~原料药的起始物料供应商应该多久被审计一次有啥规定吗?还是看API企业内部评估就可以呢?
A1:(龙马精神):对原料药的起始物料供应商的审计频次要看API企业供应商审计相关GMP文件是如何规定的。一般情况是不必每年进行一次现场审计,但当政策法规有变化可能涉及到某个供应商、对所提供的物料质量可能有影响了,就有必要进行现场审计了。但日常应对供应商资质等的变化及时关注、更新相关资料。
A2:(普):基于风险评估,一般每年都需要打分评估的,满意的得分高的供应商比如可以5年,3年再审计一次,不满意的可以每年审计一次。可参考APIC的供应商审计和管理指南。
A3:(砥砺前行):质量没有问题的,定期3年或五年,当发生质量问题,连续两批次的,可临时安排现场审计。
45、变更场地
Q:(wsf):想请教您一个问题,就是委托生产涉及到变更场地的问题,这个变更研究是由委托方指导受托方去完成呢,还是由委托方到受托方厂里去完成呢?
A1:(王冬生):没有遇到过这个问题,我理解的是品种的生产场地变更需要按变更指导原则来进行。既便生产企业帮持有人做了工作,CDE承认的依然是持有人,生产企业不能自己做变更的。
A2:(敏):肯定是持有人做啊,受托企业只负责生产,工艺变更一定要持有人审批的。
A3:(HUANG):责任肯定是持有人的。具体工作谁做,我觉得都可以。
A4:(注册圈):持有人是责任主体,负责将生产工艺转移到受托方,受托方按GMP管理规范进行工艺验证,质量研究和稳定性受托方和委托方开展都可以。
46、方法学验证
Q:(成都—RA- Lynn):请教各位老师,3类的残留溶剂能否不做方法学验证呢,从哪些角度可以论证这个点?现在的情况是在起始物料的生产中有使用,原料生产过程中没有使用,且残留溶剂并没有纳入原料注册的药典标准里面,所以能否通过ICH里面的Q3C做一个说明呢?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(江苏-RA-):方法学来自转移方,可以不做验证,只做确认。
A2:(郭美英):没有使用且不会产生,可以不做。
A3:(北京-PM/RA-Frank):CPMP/QWP/450/03 “Annex 1: specifications for class 1 and class 2 residual solvents in active substances”,去看看这.
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
你有2个论据,1个是Q3C的干燥失重,1个就是EMA对2类的要求(如果说2类都有可能不定入,那么工艺中从未用到的3类只可能比这更松).
A4:(成都—RA- Lynn):EMA也是2类.
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A5:(重庆-国际注册-蕊):2类要定的,只是说如果几个批次结果没超过限度10%,可以不用常规控制。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
47、生产地址变更
Q:(逍遥丸)集团公司的原料药登记号 ,想变更到跨省集团内的另一家公司生产,是否可以算生产地址变更呢?二家企业都是独立注册,但在股权结构上,是控股关系。
A:(吴江涛):如果是独立法人,要对方单独申报,和控股没有关系。可以走厂外车间,就算企业内部地址变更。和你们省局许可处沟通.原料药只能生产企业登记,没有委托生产的说法,技术可以来自母公司,但子公司要按照仿制要求完成全套申报资料,所有数据必须是子公司的。原料没有MAH。
48、 一致性评价
Q:(���一只大鲵哟咯咯���):各位老师好想再咨询一下,一致性评价2016年120号文中要求体内评价申报资料参照80号文要求撰写,但2020年44号文又明确了仿制药按照M4格式撰写。所以一致性评价品种的临床部分资料到底该参照80号文还是M4呢?
A1:(北京-药品注册-Mr.好先生):仿制药是仿制药,一致性评价是一致性评价.
A2:(注册圈):一致性评价品种的临床部分资料到底该参照80号文提交。
49、GMP生产设备
Q:(Ms. Vicky):我们公司计划建设一个GMP车间,打算把西林瓶水针剂和粉针剂设计在一个车间内共线生产:工艺前半段的洗瓶、烘干、罐装共用一套设备,后面根据水针剂和粉针剂工艺的不同分别用不同的设备(粉针剂将用冻干机,水针剂则不需要)。请问从法规上可行吗?是否符合最新的注册申报要求?
A:(辽宁 注册 吴文哲):可以没有问题,这其实也不涉及到共线。而且本身的西林瓶都是一次性使用的。灌装的针头应该也不会交叉使用。我们抗体车间也是那么设计的,配备自动进出料系统即可。GMP设施章节,其中明确说明了 设备多产品共用的可行性,并有相应评估报告 也就是说你必须得做风险评估,具体还得根据结合你的产品来,在GMP法规中没有禁止这种行为。风险评估报告,在厂房建设阶段的话是不需要的,后面NDA/BLA的时候需要有。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
50、文件核酸检测
Q:(简):大家好, 寄送药用辅料的给国家局也要做核酸检测和消毒的吗?
A1:(菡雪儿):我上周交的光盘资料,是低风险地区,没有做核酸,接受了,现在还没有接到异常情况反馈呢。
A2:(特雷西):按CDE在1.29号的通知中填写申报资料明细表并发送指定邮箱,如果没有境外资料并低风险地区寄出,是不用做核酸的。到时候业务处老师会按照邮箱中发送的明细表信息核对签收快递。
文章内容出自注册圈交流群~欢迎大家进群交流学习,有解答不到之处 还请斧正
|



 狗仔卡
狗仔卡
 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 变色卡
变色卡 抢沙发
抢沙发 千斤顶
千斤顶 显身卡
显身卡