F.DOCUMENTATION RELATING TO COMPLIANCE WITH GOOD MANUFACTURING PRACTICE(GMP) FOR THE INVESTIGATIONAL MEDICINAL PRODUCT(研究用药品符合药品生产质量管理规范(GMP)的相关文件)
G. INVESTIGATIONAL MEDICINAL PRODUCT DOSSIER (IMPD)(试验用药品申报资料)
H. AUXILIARY MEDICINAL PRODUCT DOSSIER(辅助药品申报资料)
I. SCIENTIFIC ADVICE AND PAEDIATRIC INVESTIGATION PLAN (PIP)(科学建议和儿科研究计划)
J. CONTENT OF THE LABELLING OF THE INVESTIGATIONAL MEDICINAL PRODUCTS(试验用药品标签的内容)
PART II:
K. RECRUITMENT ARRANGEMENTS (information per member state concerned)(招募安排(成员国关注的信息))
L. SUBJECT INFORMATION, INFORMED CONSENT FORM AND INFORMED CONSENT PROCEDURE (information per member state concerned)(受试者信息、知情同意书和知情同意程序(成员国关注的信息))
M. SUITABILITY OF THE INVESTIGATOR (information per member state concerned)(研究者的适用性(成员国关注的信息))
N. SUITABILITY OF THE FACILITIES (information per member state concerned)(设施的适宜性(成员国关注的信息))
O. PROOF OF INSURANCE COVER OR INDEMNIFICATION (information per member state concerned)(保险凭证封面或赔偿(成员国关注的信息))
P. FINANCIAL AND OTHER ARRANGEMENTS (information per member state concerned)(财务和其他安排(成员国相关信息))
Q. PROOF OF PAYMENT OF FEE (information per member state concerned)(费用支付证明(相关成员国信息))
R. PROOF THAT DATA WILL BE PROCESSED IN COMPLIANCE WITH UNION LAW ON DATA PROTECTION(将按照数据保护联邦法律处理数据的证明)
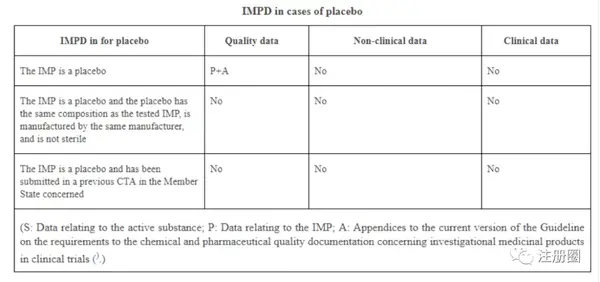
重点阐述IMPD文件:撰写格式可参考EMA法规:
Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials.



 狗仔卡
狗仔卡


 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 变色卡
变色卡 抢沙发
抢沙发 千斤顶
千斤顶 显身卡
显身卡