交流群问答集锦 -2022(8.22-8.26)
01工艺验证
Q:(广东注册Chloe):原料药的关键起始物料有两个供应商,合成路线不一样,工艺验证是否需要用两个供应商的起始物料去做,各做三批还是一共三批即可?
A:工艺验证和稳定性分别做连续三批是最推荐的。行业里之前也有验证前两批用厂家A的SM,后一批用厂家B的SM,这要看审评人员的尺度和大环境,审评也是与时俱进的,过去的经验只能供参考。另外一种方法是,NDA时只用厂家A的SM,后面获批后新增SM的供应商,这个还是较好操作的。
02原料药变更
Q:(北京进药品注册-赵宁宁):我们买了一个仿制药品种,但是该品种的原料药停产了。我们计划自己做原料药,然后对制剂做补充申请,对原料药进行变更。这种情况下,我们变更后原料药的资料可以放在补充申请里直接提交吧?还是需要对原料药进行登记,补充申请进行关联(变更后的原料药未批准)?
A1:(上海项目管理-卢峰):看您们对原料药后期市场是怎么规划:
1)若原料药只供应自己的制剂,补充申请就可以。
2)若原料药考虑后期做市场推广销售,建议这种仿制原料药就可以登记申请单独技术审评,单独技术审评通过有利于后期市场推广。
A2: 无论是1)或者2),都要评估变更原料药生产商后,对制剂关键性能的影响,必要时要做相应研究。
03技术转移
Q:(谷):我们是甲方,全权委托乙方对我们的新药项目进行研发,那么药物研发技术转移方案和报告,是不是由乙方来撰写提交给我方?这个转移包括工艺和分析两部分是吗?是分开写还是放到一个方案中?
A1:(成都 CMC&RA):通常由企业TTD来组织技术转移,分转移方和接收方,职责划分通常转移方起草方案,完成转移后接收方完善转移报告,至于要分开还是放一个方案都可以。符合企业技术转移管理规程就行。
A2: 如果甲方对转移前乙方的所有研发工作十分熟悉,可以由甲方主导技术转移工作,甲方告知乙方准备相应的转移文件、对照品、耗材等;指导技术转移全过程,审核转移方案和报告等。
04工艺验证
Q:(豫小南):有一原料药申请了单独审评,审评费已缴,目前正在审评中。现有一制剂使用该原料药,可以直接用注册批的原料药做制剂工艺放大吗?如果可以用的话,万一制剂还没有申报,原料药现场核查了有影响吗?还有这个原料药是只能以赠送的方式提供给制剂厂家,还是也可以以销售的方式提供,合同中注明仅供制剂研究用。
A:(广东 药品研发):可以用原料药工艺验证的样品做制剂的工艺验证或者制剂的放大批,不过要评估原料药审评不过的风险,其实就是原料药+制剂的研发模式,正常采购,原料药厂家在合同里会注明研发用。
05杂质研究
Q:(豫小南):目前在做一个注射剂,其原料药没有做元素杂质研究,而制剂在工艺组件、包材相容性试验中把1、2A、3类元素杂质都有检测,如果该部分元素杂质符合要求,那还需要提供原料药的元素杂质研究吗?
A1:(京-打雜):原料药的元素杂质来源跟制剂的不可能完全一样的, 是需要对API的元素杂质进行研究和评估的。
A2: 如果原料杂质评估后,可能引入的元素杂质含在制剂研究的元素杂质之中,可以不提供原料药的元素杂质研究,但要有原料药元素杂质评估报告。
06审评问询函
Q:(wj):CDE给公司发的“药学专业审评问询函”,让我们在5个工作日内逐条回复每一项需要补充的内容,问题是要补充实验、更改好些内容,在这几天内也完成不了啊。提出的那些建议,我们就是要研究的;他都提出来了,我们咋回复啊?
A1:(沪-打杂-��� ):第二章 专业审评问询
第五条 药审中心在专业审评期间或综合审评期间,专业主审或主审报告人在充分审评基础上对申报资料有疑义或认为内容存在问题,经审评部门负责人审核后,通过药审中心网站向申请人发出“专业审评问询函”,告知申请人存在问题的具体内容、依据和要求等,并要求在5个工作日内进行解释说明或书面回复。
审评部门在审评过程中对需要发补的问题应发送“专业审评问询函”提前告知申请人。但“专业审评问询函”并不是正式书面补充资料通知,也不代表最终审评决策意见,审评计时不暂停。
第六条 药审中心通过“专业审评问询函”告知申请人以下信息:
1)无需开展研究即可提供的证明性材料;
2)不需要补充新的技术资料,仅需要对原申报资料进行解释说明;
3)审评认为可能需要补充完善的缺陷问题。
第七条 申请人应在“专业审评问询函”发出5个工作日内进行解释说明或书面回复。对于需要书面回复的,申请人应在5个工作日内进行电子提交,同时在时限内寄出与电子版一致的纸质版资料,通过药审中心网站下载打印“专业审评问询函”作为接收补充资料及纳入档案的依据。
第三章 正式发补、发补咨询和异议程序
第八条 在审评过程中需要申请人在原申报资料基础上补充新的技术资料的,结合“专业审评问询函”的答复情况,根据《药品注册管理办法》规定,药审中心原则上提出一次补充资料要求,列明全部问题后,以书面方式通知申请人在80个工作日内补充提交资料。
基本上应该老师是觉得你们80工作日补不上来,先给你们打个预防针
A2:(中美双报-人药-heheng):我们前面收过一次专业问询号,基本上就是后面的完整发补内容。提前告知了。
A3:(广东注册Chloe):我们有在发补后收到这个问询函,联系了老师,说超过5个工作日也没关系。
07仿制药申报
Q:(广东注册 Chloe):4类仿制药,口服常释固体制剂,在国内外上市多年,需要做非临床和临床吗?
A1:(注册er):不需要。
A2: 一般不需要做非临床,除非你有超过界定限的杂质;要考虑是否可以BE豁免,否则要做生物等效性试验。
A3:非临床和验证性临床不需要,生物等效性需要评估能否豁免。
08药典备案
Q:(北京 公共事务注册 wen):药典备案时,当企业注册标准和药典标准不一致时,省局要我们按CP2020要求进行备案,这个有相应的指导原则么?
A:(京-打雜-���):你查查那个药典发布的文。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
按这条,你标准低的要按药典,就得走变更。变更注册标准起步就是中等。CDE有已上市化学药品变更指导原则(2021年)。因为你的标准低于药典,所以要执行药典。而提升标准需要做注册标准变更,收严是中等,所以是备案。
09标准变更
Q:(山东 注册 程程):审评过程中仅收紧限度但方法不变,此类标准变更是否需要省所复核?
A:(上海 注册 贰雯):收紧限度,之前的复核结果仍在变更后限度内的,可以不再经过省所复核。但是建议先沟通。我们之前也遇到过这类问题,但还是会先电话问审评老师意见,一般不用再检测。然后带着CDE意见问省所有没有额外的流程需要走,比如省所是否需要出具其他类型的报告。
10疫苗批次
Q:(***):疫苗特性鉴定、结构确证,是需要做3批吗,有法规依据吗?
A:(星星):建议3批。
11非临床试验
Q:(任霞):4类注射液的非临床试验研究,溶血、刺激性、过敏试验的实验结果是否写在资料的2.4非临床综述里?
A:(南京 注册 飞飞):2.4和2.6都要写,报告要在模块4提交。
12注册检验
Q:(浙江 RA Victoria):进口原料药注册检验,三倍的样品和对照品必须要分装么?可不可以是一个批次装一瓶?
A:(吴艳平):每个批次需要按照检验项目分装多个小瓶,比如10个检测项目,每批单倍量最好分装11个小瓶,最少的分装量也要按照理化、液相、气相、元素杂质等检测项目分装。对于原料药,一个批次装一瓶肯定不行。至于装几瓶还是看自己品种的情况。我说的就是一个大原则最大分装量和最小分装量。
13风险管理计划
Q:(若椰溪 北京 注册 ):申请IND时,模块1的1.8.3风险管理计划要提交吗?
A:(浅山):交;之前受到过发补,要求补充。
https://www.cde.org.cn/main/news/viewInfoCommon/77e34e30c7141b2770ddd6f80e80f9ff
14委托生产
Q:(广州-注册-沸沸):未来 A公司将作为MAH ,但是现在没有建立相关的质量体系等,在I期II期的时候可以委托其他企业生产吗中试或者II期批的样品吗?就是他这个委托的前置条件是什么呢?
A1:(上海-注册-Rachel):可以委托,但需要建立MAH的质量体系。最好在工艺验证前完成MAH质量体系的搭建,后续还要拿B证。
A2:(Sword):做好质量审计,双方要签署质量协议。
15补充申请资料
Q:(上海-注册-小草):临床试验期间方案变更走补充申请的话,5.药学研究资料,6.药理毒理研究资料,7.临床研究资料需要按照CTD格式整理吗?
A:(上海-注册-xwfunny):可以参考CTD资料的结构。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
16资料格式
Q:(HN RA Henry):国内注册药典格式的质量标准需要放在3.2.P.5.1吗?还是模块1的1.3.3放了就可以了呢?有没有哪个文件有明确规定的呢?
A1:( 北京-注册-吴正宇 ):当搞不清楚鸡蛋放哪个篮子里时,可取的做法是都放。
A2: 一般情况下,1.3.3是要放的,P.5.1正文可以不放。
17储存温度
Q:(广州-RA欣):如果长期稳定性做了25℃的条件,那药品的储存温度可以是室温吗?
A:( 成都-质量研发 ):美国是可以的,但中国不行。中美虽然都是II气候带,但年平均温度不同,你只有在30℃下考察了长期试验,才能在中国市场上说是室温保存。参考ICH Q1。
18氮气分类
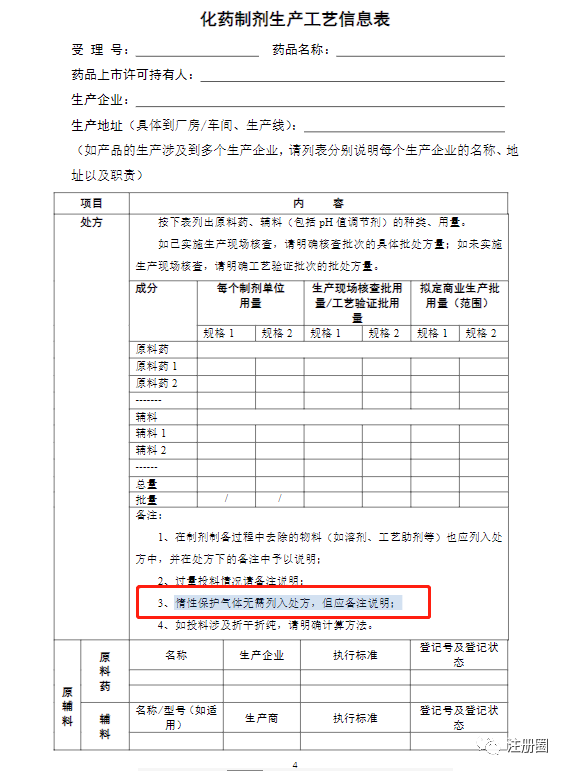
Q:(上海-药品注册-周小碗):制氮机制备的氮气是否需要按照辅料管理呀?有没有哪个法规有提到呢?这个氮气是注射剂生产工艺中充氮作为保护气体的。
A1:(RA-郭星星):生产工艺信息表模板有提这个,另外,不列入说明书,不写入申请表。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
A2:(钟无语):氮气不算辅料,可以看下《化学药品生产工艺、质量标准通用格式和撰写指南》,但是氮气需要符合要求。应根据剂型等建立适合品种的标准。
A3:(北京-CMC-台海隧道):使用后进入人体算原辅料,仅用来保护活性成分,使用前排掉了不算。
19细菌内毒素
Q:(青岛RA_Arya):药典上对眼用制剂没有进行细菌内毒素的规定,生产眼用制剂的辅料需要限定细菌内毒素吗?
A:(Sword):从目的反推手段,既然产品没有内毒素的要求,那么辅料自然也没有内毒素要求。
Q:(青岛RA_Arya):但是在GMP里面提到无菌药品的辅料和包材需要进行细菌内毒素控制呢?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(Sword):这段话的重点在于安全性,影响安全性的质量指标,需要检验。
20原材料变更
Q:(南京-注册-Cherry):在临床试验期间,变更原材料,将非GMP级别的变成GMP级别的,属于中等变更吗,有什么法规可以参考吗?
A1:(Sword):个人理解,这属于微小变更。按照CDE指南的意思,凡是行动过后加强了对产品安全性、有效性、质量可控性的控制力度的,都属于微小变更;相反,控制力度减弱的,属于中等变更,有失控风险的,属于重大变更,这段话的依据是《已上市化学药品药学变更研究技术指导原则(试行)》,没记错的话是2021年年底发布的。这里面参考的内容是,两个截图是《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》的内容。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A2:(张新春 江苏 上药常药):看原材料是API 还是辅料,还是变更生产厂家,还是同厂家工艺。
A3:需要根据变更原材料的作用性质进行综合评估,且需要评估非GMP级别的变成GMP级别二者关键质量属性的差异。
21药品专利
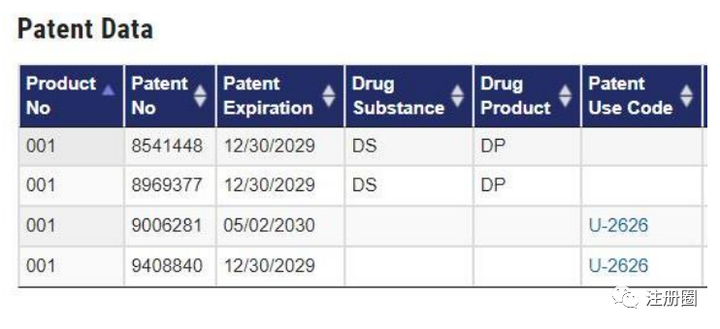
Q:(山东-注册-anne):这四个专利在中国同族专利为同一个,为什么在美国要申请4个专利号啊?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(武汉-运营-温辉):今年5月29日《药品管理法实施条例》有提及数据保护,供参考。征求意见稿。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
22细胞来源
Q:(北京-QC-王富贵):申报批的细胞都哪些可以外包?毒理?三批工艺验证及检测方法学验证?
A:(宜明细胞刘双生):都可以。
23安慰剂资料
Q:(上海-进口注册):生物制品的临床试验申请,安慰剂的资料是跟IND一起交的吗?有没有IND时不交,后面被发补的情况?因为安慰剂这块资料没做好,其他的资料都好了,想着能不能先递交试验药的资料,安慰剂的后面发补了交。
A1:(图图菊):遇到过,只能补充5日内能说明说清的问题。
A2: 安慰剂的资料一般是跟IND一起交,如果真交不上又想抢时间,可以尝试等问询时5日内交上,风险自担。
24精密度RSD
Q:(山东–注册–vivian):HPLC测有关物质时,精密度RSD要求需要严格按照药典中的要求执行么?分析部门的同事给出RSD 限度都高于药典要求。这样申报没问题么?
药典中分析方法验证指导原则:
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(成都-注册&CMC-Szechwan):一般需要严格按照药典执行,但是对于限度小的,RSD可以适当放宽。报国内就严格按药典和ICHQ2 来。如果报欧美可以justify,就是证明自己能达到的重复性已经是目前技术最好的状态,同时也满足质控要求。
25适应症变更
Q:(Nicole):5.1类进口药品注册,国内申请的适应症与参考国的适应症不一样(国内的少),受理会有什么问题吗?
A:(注册圈):如果国内申报的适应症是国外申报适应症涵盖的,这么申报受理没有问题,批准的适应症只能是国内申请的适应症。
26工艺合并
Q:(大彬):一个颗粒剂产品,两个等比规格仅装量不同,能否将灌装前的工序合并?比如拟申报批量均为10万袋,10mg规格和15mg规格10万袋的批重量分别是50kg和75kg,是否可以将罐装前的工艺合并进行,如进行125kg的罐装前工序,最后再分别罐装10mg规格10万袋和15mg规格10万袋?这样相当于罐装前的工艺批量比拟申报的批量要大,大批量验证通过是否可以说明小批量的验证也是通过的?比如批准后这两个规格可能会根据市场需求分别生产。或者对罐装前的工艺分别按两个亚批进行连续生产是不是会更好一点?这样亚批的批量和实际批准后生产的批量就是一致的。
A:(注册圈):工艺验证是全工序验证,不同装量规格包装工序也需要验证。
27补充申请
Q:(乐多):原料药的补充申请事项下的重大变更是按直接变更途径申报还是按照补充申请项下的。
A:(注册圈):补充申请。
28稳定性研究
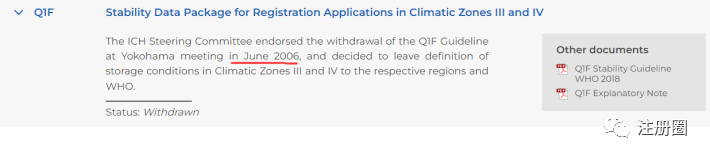
Q:(高):药品出口东南亚国家,稳定性研究还能用ICH Q1F这个指南吗?
A:(注册圈):ICH Q1F <Stability Data Package for Registration Applications in ClimaticZones Ill and lv>已于2006年6月废止。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
29参比制剂
Q:(广州-注册-hard candy):一个品种参比制剂目录公布原研进口和美国上市两个来源的的都可以选为参比,但是原研进口中有一个规格国内买不到了,可以一个规格买国内参比一个规格买美国上市的参比吗?
A:(注册圈):法规上没问题,实际操作过程中要考虑国内参比制剂和美国上市的参比制剂不同产地参比制剂之间质量的差异性。
30原辅包备案
Q:(江苏注册271):状态为I的辅料要转A的话,能不能通过制剂的辅料供应商变更去完成?还是只能通过新药或仿制药申报?
A:(注册圈):只能通过新药或仿制药申报。
31方法变更
Q:(注册-秋):对于质量标准方法变更,省所方法复核时,需要提供几批样品?是已上市产品。
A:(注册圈):三批样品
32申请人变更
Q:(大脸猫大脸猫爱吃鱼):生物类似药临床1期已完成,3期试验还未开展,变更申请人应该是什么流程?
A:(注册圈):直接变更,变更实施后,申办者需要按照相关要求在药物临床试验登记与信息公示平台更新信息,并在《研发期间安全性更新报告》(DSUR)中汇总报告。
33IND申请
Q:(西山玉湖):新药IND申请,申请的是大适应症,只提交了1期方案,之后要做1b或2期的时候,还需要提交补充申请吗?
A:(注册圈):没有重大变更一般是不需要的。
|



 狗仔卡
狗仔卡
 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 变色卡
变色卡 抢沙发
抢沙发 千斤顶
千斤顶 显身卡
显身卡