为鼓励新药创制,严格审评审批,提高药品质量,促进产业升级,NMPA在2016年51号文对当前化学药品注册分类进行改革,明确原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。定义5.1类为境外上市的原研药品(包括原料药及其制剂)申请在境内上市。本文对进口原研药品注册路径和策略进行深入分析解读,并通过案例分享,总结在制定申报策略时需主要考虑的因素,供大家参考。
进口原研药品注册路径和策略
路径1:境外原研进口,按1类或2类药品国内外同步研发申报注册策略:自临床早期研发阶段即参与全球同步研发。
- 中国受试者参与首次人体试验成为新趋势
- 在II期POC阶段加入,为III期开发积累中国数据(依据适应症寻求II期数据加速批准的可能)
- 加入国际多中心III期试验
对于首个适应症如果没能加入全球:可考虑纳入香港、台湾和亚洲国家,为后续中国早日上市布局。
路径2:境外原研进口,若境外已上市境内未上市,按5.1类首次申报注册策略:
5.1首次在境内申报
- 有中国人群PK和/或PD、有效性安全性数据的药品, 获益大于风险,相关境内外数据可直接支持上市申请
- 无中国人群数据,且研究未见明显种族敏感性范围:严重或危及生命疾病、罕见病且无有效治疗手段的药品,或较现有治疗手段有明显提高疗效或安全性等优势结果:附条件批准,需开展上市后安全、有效性研究
5.1境内上市后的LCM
- 增加境外已批准境内未批准的新适应症:申报前与监管机构沟通,有豁免中国临床试验可能。
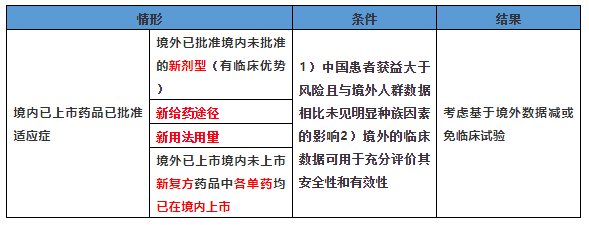
- 用于境内已上市药品已批准适应症:
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
- 缺乏种族敏感信息或存在种族敏感性:应在上市前开展相关桥接性临床试验
路径3:境外原研进口,若原研境内未上市,仿制药先上市,按5.1类申报注册策略:需具体问题具体分析,加强与监管机构沟通交流
- 仿制药通过一致性评价,CDE认可仿制数据,则可尝试与CDE沟通豁免中国临床
- 如果已上市仿制药数据不足,则可能需要补充必要的桥接数据
路径4:进口原研产品境内上市前地产化注册策略:
- 早期试验IND注册境外产地,在III期前转移至境内生产,Ph3 IND申请境内MAH和境内产地
- 上市申请国内、外同步递交(须在国外批准前递交)
- 在境外批准后递交:注册分类3类,申请递交和批准的复杂性
路径5:进口原研产品境内上市后转地产,目前路径尚不明确
案例分享
1. 阿帕他胺:原研药品5.1申请免临床有条件批准上市,急需名单药品
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
2. 盐酸托莫西汀口服溶液:境外已批准境内未批准的新剂型(2018年获批),豁免中国BE上市
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
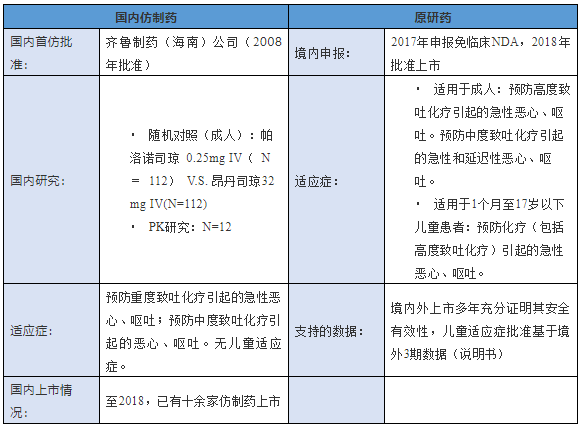
3. 盐酸帕洛诺司琼注射液:境内仿制药先上市,境外原研豁免临床上市同时豁免境内未批准儿童适应症
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
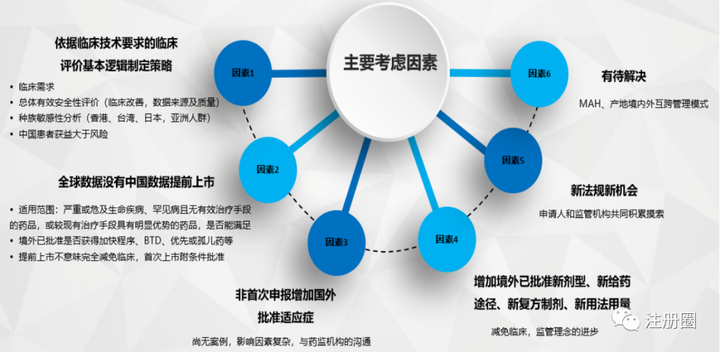
注册策略主要考虑因素
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
相关参考法规
1.《药品技术转让注册管理规定》(国食药监注〔2009〕 518号)(2009-08-19)
2.《成人用药数据外推至儿科人群的技术指导原则》 2017年79号文(2017-05-18)
3.《接受境外临床试验数据的技术指导原则》 2018年第52号文(2018-07-10)
4.《临床急需境外新药审评审批工作程序》 2018年79号文(2018-10-30)
5.《境外已上市境内未上市药品临床技术要求》 2020年第29号文(2020-10-16)
6.《已上市药品临床变更技术指导原则(征求意见稿)》(2020-04-30)
7. ICH E5:接受国外临床试验数据的种族因素
8. ICH E17:多区域临床试验计划和设计的一般原则
文章自注册圈公众号~欢迎关注哦~
|



 狗仔卡
狗仔卡
 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 变色卡
变色卡 抢沙发
抢沙发 千斤顶
千斤顶 显身卡
显身卡